Understanding Cystic Fibrosis
Cystic fibrosis, also known as CF, is an inherited genetic disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, causing the CFTR protein to malfunction.
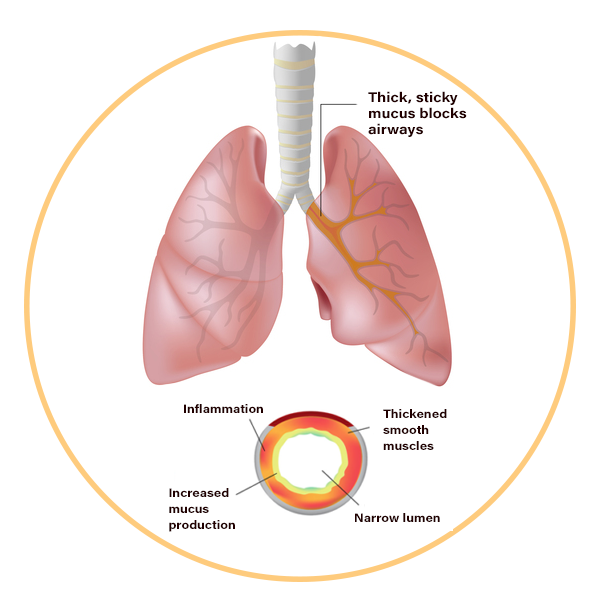
When the protein is not working correctly, it’s unable to help move chloride to the cell surface. Without the chloride to attract water to the cell surface, the mucus in various organs becomes thick and sticky. Instead of acting as a lubricant, the secretions plug up tubes, ducts and passageways, especially in the lungs and pancreas.
Although cystic fibrosis requires daily care and treatments, people with cystic fibrosis are usually able to attend school and work. Improvements in screening and treatment options have given people with cystic fibrosis the ability to live into their mid- to late 30s, on average, and some are living into their 40s and 50s.
What Causes Cystic Fibrosis?
Cystic Fibrosis is a genetically inherited condition. For someone to have CF, they need to inherit a mutated gene from both of their parents. For couples who want to have children, genetic testing is also important as more than 10 million Americans are carriers of a CF gene. For every pregnancy, there is a one-in-four chance that the child will have CF when both parents are carriers.
People with only one copy of the mutated gene are called carriers. They do not have the condition or any of its symptoms. To have the disease, both parents must be carriers of the mutated gene.
If two carriers have a child, there is a:
- 25% chance the child will have CF
- 50% chance the child will be a carrier but will not have CF
- 25% chance the child will not be a carrier and will not have CF

Signs & Symptoms of Cystic Fibrosis
Signs and symptoms of cystic fibrosis vary depending on the severity of the disease. Most of the symptoms of cystic fibrosis affect the respiratory system and digestive system. The thick and sticky mucus associated with cystic fibrosis clogs the air passages that carry air in and out of your lungs as well as block tubes that carry digestive enzymes from the pancreas to the small intestine. Without these digestive enzymes, the intestines are not able to fully absorb nutrients from food.
- Salty-tasting skin
- Persistent cough
- Recurring lung infections
- Wheezing, shortness of breath
- Wheezing
- Poor weight gain or growth
- Greasy, bulky stools
- Nasal polys
Airway Clearance Therapy for Cystic Fibrosis
In order to clear their airways, people with Cystic Fibrosis typically perform Airway Clearance Therapies (ACTs) daily, sometimes several times a day. This can be done techniques such as active cycle of breathing, manual chest physiotherapy (CPT), a high frequency chest wall oscillation (HFCWO) therapy vest, or a PEP device. These methods use percussion, vibration and drainage to help loosen and mobilize mucus in the airways, allowing it to be coughed up.